Om DNA-sekvenser, transkripsjonsfaktorer og motivoppdaging
Proteiner, gener og DNA
Proteiner er en gruppe store molekyler som utgjør kanskje de viktigste komponentene i levende organismer.
Selv om proteiner kan virke store og kompliserte, er de bygget opp på en veldig enkel måte.
Hvert protein er i utgangspunktet bare en lang kjede bestående av ulike aminosyrer.
Det finnes 20 forskjellige typer aminosyrer og rekkefølgen av disse i kjeden bestemmer
hvilken form et protein vil få og hvilke egenskaper det vil ha.
Mange proteiner brukes som byggesteiner i kroppen, slik som i hår, negler og muskelfibre.
Den aller viktigste rollen til de fleste proteiner er likevel å
fungere som enzymer – katalysatorer for kjemiske reaksjoner. Knapt noen ting
foregår i en celle uten at enzymer på en eller annen måte er involvert i prosessen. Enzymer
bryter ned molekyler og bygger opp nye, de flytter rundt på kjemiske grupper fra et molekyl
til et annet for å endre de kjemiske egenskapene, og de utvinner energi fra næringsstoffer.
Enzymer kan også skape målrettet bevegelse,
både på mikroskopisk nivå innenfor en enkelt celle og på organisme-nivå når flere enzymer aktiveres
samtidig. Hvert enkelt enzym har sin egen spesifikke oppgave som den utfører på en kontrollert måte.
I mennesker finnes det over hundre tusen mer eller mindre forskjellige typer proteiner,
og nye proteiner synteseres kontinuerlig i alle levende celler.
Prosessen med å sette sammen nye proteiner foregår selvsagt også ved hjelp av enzymer (som alt annet).
For at apparatet som bygger proteiner skal kunne klare denne oppgaven, trenger de tilgang på oppskrifter som
spesifiserer hvordan hver type protein skal bygges opp. Denne oppskriften
på proteiner finner man i genene.
Et gen er en bit av et DNA-molekyl som inneholder
oppskriften på hvordan en type proteiner skal lages
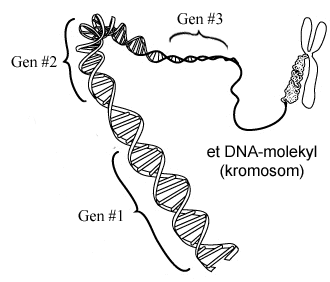
DNA (deoxyribosenukleinsyre) er altså det molekylet som inneholder den genetiske oppskriften.
Samtlige celler i en organisme inneholder en fullstendig kopi av all DNA-informasjon.
På samme måte som proteiner er også DNA-molekylet bygget opp som en lang kjede av de samme komponentene,
men DNA består ikke av aminosyrer men derimot av fire ulike nukleotider angitt som A, C, G og T.
Et gen er en liten bit av DNA-molekylet som igjen svarer til en bestemt sekvens av de fire nevnte
bokstavene. Koblingen mellom DNA-sekvensen og oppbyggingen av proteiner er enkel og genial:
Hver gruppe på tre bokstaver i DNA-sekvensen koder for en bestemt aminosyre i proteinet. Eksempelvis
vil sekvensen "ACC" i et gen bety at aminosyren "Threonine" skal settes inn i neste posisjon i protein-kjeden.
Sekvensen "ATGGGCGGT" leses som "ATG-GGC-GGT" og oversettes til en sekvens av aminosyrene Methionine-Glycine-Glycine.
(Siden de fire bokstavene A,C,G og T kan danne "ord" på tre bokstaver på 64 ulike måter,
mens det bare finnes 20 forskjellige aminosyrer, kan det være at flere ulike ord spesifiserer samme aminosyre slik
som GGC og GGT her). En gen-sekvens som er 300 nukleotider lang vil derfor spesifisere et protein som består av 100 aminosyrer.
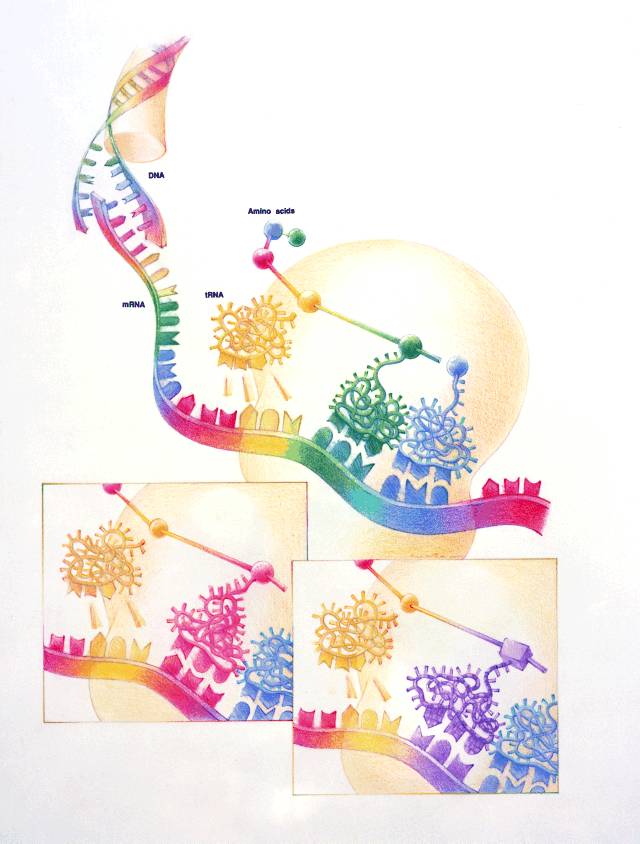 Når et protein blir produsert fra et gen sier vi at genet uttrykkes. Prosessen med å uttrykke et gen foregår
hovedsakelig i to trinn. Første steg kalles transkripsjon og går ut på at det lages en arbeidskopi av DNA-sekvensen til genet.
Denne kopien (kalt mRNA) fraktes så til ribosomene som utfører arbeidet med å sette sammen
proteinet i henhold til oppskriften. Dette siste steget i prosessen kalles for translasjon siden nukleotid-sekvensen
fra DNA "oversettes" til en sekvens av aminosyrer.
Når et protein blir produsert fra et gen sier vi at genet uttrykkes. Prosessen med å uttrykke et gen foregår
hovedsakelig i to trinn. Første steg kalles transkripsjon og går ut på at det lages en arbeidskopi av DNA-sekvensen til genet.
Denne kopien (kalt mRNA) fraktes så til ribosomene som utfører arbeidet med å sette sammen
proteinet i henhold til oppskriften. Dette siste steget i prosessen kalles for translasjon siden nukleotid-sekvensen
fra DNA "oversettes" til en sekvens av aminosyrer.
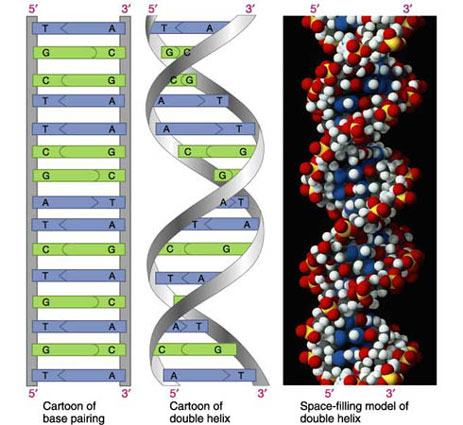 Selv om den genetiske oppskriften kan defineres ved å bruke kun én sekvens av nukleotider,
er DNA-molekyler gjerne satt sammen slik at to nukleotid-sekvenser er koblet opp mot hverandre.
Hver nukleotid i den ene sekvensen er da koblet til et nukleotid i den andre sekvensen på en helt bestemt måte,
nemlig ved at en A i den ene sekvensen parrer seg med T i den andre (og tilsvarende med G og C).
De to sekvensene er derfor ikke uavhengige av hverandre, og hvis man vet hvordan sekvensen ser ut på den ene siden,
kan man lett resonere seg frem til hvordan den ser ut på den andre siden.
De to nukleotid-sekvensene roterer også om sin egen akse langs molekylet slik at de danner en spiralform
(eller en "dobbel helix" som det heter på fint).
Selv om den genetiske oppskriften kan defineres ved å bruke kun én sekvens av nukleotider,
er DNA-molekyler gjerne satt sammen slik at to nukleotid-sekvenser er koblet opp mot hverandre.
Hver nukleotid i den ene sekvensen er da koblet til et nukleotid i den andre sekvensen på en helt bestemt måte,
nemlig ved at en A i den ene sekvensen parrer seg med T i den andre (og tilsvarende med G og C).
De to sekvensene er derfor ikke uavhengige av hverandre, og hvis man vet hvordan sekvensen ser ut på den ene siden,
kan man lett resonere seg frem til hvordan den ser ut på den andre siden.
De to nukleotid-sekvensene roterer også om sin egen akse langs molekylet slik at de danner en spiralform
(eller en "dobbel helix" som det heter på fint).
I de fleste organismer er genene fordelt over flere separate DNA-molekyler, og hvert av disse
kalles da for et kromosom. Mennesker har for eksempel 46 kromosomer, hvor halvparten er arvet
fra far og halvparten fra mor. Den samlede mengden genetisk informasjon i en organisme kaller man for
organismen genom. Det menneskelige genomet består av rundt 3 milliarder nukleotider.
Hele denne sekvensen er nå blitt kartlagt og gjort tilgjengelig i databaser.
Nå er det overhodet ikke slik at alle deler av et DNA-molekyl består av gener. Faktisk er det estimert
at så lite som 2% av det menneskelige genomet utgjøres av slike protein-oppskrifter. Hva resten av DNAet brukes til
har man fortsatt ikke helt oversikt over. Store deler har tilsynelatende ingen nyttig funksjon og
ble i tidligere tider gjerne omtalt som "junk DNA". Etterhvert som forskningen gjør fremskritt har man
dog oppdaget at mange slike "ikke-protein-kodende" deler faktisk har viktige oppgaver likevel.
En del av DNA som spiller en svært viktig rolle er de cis-regulatoriske regionene.
Dette er biter av DNA-sekvensen som er med på å styre hvordan gener blir uttrykt.
Regulering av gen-uttrykk
Et menneske er et eksempel på en komplisert organisme bestående av mange organer som igjen
er bygget opp av ulike celletyper. Muskelceller, hudceller, blodceller og hjerneceller
har helt klart forskjellige utseende og egenskaper, og som vanlig er det proteinene som bestemmer
disse egenskapene i hver celle. Ulike celler inneholder nemlig forskjellige typer proteiner og
i forskjellig mengde.
Noen proteiner produseres stort sett i alle celler, slik som proteiner som deltar i transkripsjons-
og translasjonsprossesen av gener. Andre proteiner dannes derimot kun i spesialiserte
celletyper. Eksempelvis produseres proteinet insulin utelukkende i celler i bukspyttkjertelen.
Likevel er det slik at samtlige celler i kroppen inneholder nøyaktig den samme genetiske informasjonen.
Inne i hver cellekjerne finnes det en fullstendig kopi av alle de 46 kromosomene.
Det som skiller de ulike cellene fra hverandre er derfor ikke hvilke gener de innholder men derimot
hvilke gener de uttrykker.
Celler uttrykker heller ikke de samme genene hele tiden, men produserer forskjellige
proteiner etter hvert som behovet for dem oppstår. Når en celle skal dele seg må den gå gjennom mange trinn,
og i hvert trinn er det bestemte proteiner som utfører oppgavene før andre proteiner tar over jobben i neste trinn.
For at en organisme skal kunne utvikle seg riktig fra en befruktet eggcelle til et voksent individ, og
ellers fungere normalt under alle forhold, er det helt avgjørende at rett gener uttrykkes på riktig tid og sted.
Cellene har derfor mange mekanismer som er med på å regulere hvordan gener uttrykkes basert på indre eller ytre signaler.
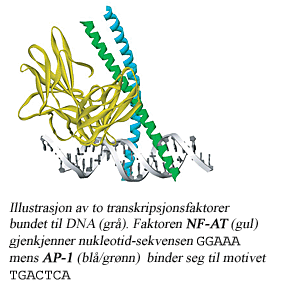
En viktig del av dette gen-regulatoriske systemet utgjøres av de såkalte transkripsjonsfaktorene.
Dette er proteiner som binder seg til DNA-strengen, vanligvis i området som ligger foran et gen,
og dermed medvirker til enten å aktivere eller blokkere transkripsjon av genet.
Delen av DNA-sekvensen som ligger foran genet kalles gjerne for en promotor og er et eksempel på en cis-regulatorisk region.
Hver enkelt transkripsjonsfaktor gjenkjenner
en bestemt nukleotid-sekvens i DNA-kjeden, et bindingsmotiv, som den fester seg til.
Selve det fysiske området hvor faktoren binder kalles for et bindingssete. En promotor-region inneholder
gjerne bindingsseter for flere forskjellige transkripsjonsfaktorer, og gen-uttrykket avhenger da
av hvilke av disse faktorene som er tilstede i cellen og kan binde seg til enhver tid.
Et mål for biologer og medisinere er å få oversikt over hvordan alle genene i en organisme reguleres.
Et viktig ledd i dette arbeidet blir da å identifisere hvor i genomet ulike bindingsseter er lokalisert,
siden dette kan gi svar på hvilke faktorer det er som styrer uttrykket av de enkelte genene.
Slike bindingsseter og motiver kan (og strengt tatt må) identifiseres ved hjelp av laboratorieforsøk.
Problemet er at dette både er tidkrevende og dyrt, for man kan bare undersøke små DNA-biter av gangen.
(typisk noen hundre nukleotider). Med 3 milliarder nukleotid-par i det humane genomet,
sier det seg selv at det vil ta langt tid om man skal undersøke alt manuelt under alle forhold.
Det er her bioinformatikk og automatisk motivoppdaging kommer inn.
Ved hjelp datamaskiner og statistiske analyser kan man identifisere deler av DNA-sekvensen
som er mer sannsynlige kandidater for å være bindingsseter, og kanskje til og med si noe om
hva slags faktorer det er som kan binde til disse setene. Slik informasjon vil igjen
muliggjøre en mer fokusert tilnærming til arbeidet med å bekrefte bindingsseter empirisk.
|
 |
Utdrag fra 10 promotor- sekvenser hvor bindingsseter for to
ulike transkripsjons- faktorer er angitt. TATAA-motivet bindes
av faktoren TBP mens Sp1 binder til den såkalte "GC-boksen"
GGGCGGGC
(Artistisk illustrasjon!)
|
Automatisk motivoppdaging
Hittil er det gjort lite forskning med hensyn på å avdekke hvordan
slike bindingsmotiver bør se ut, for eksempel om det er visse egenskaper
ved ulike nukleotid-sekvenser som gjør at noen er gode kandidater for å
binde transkripsjonsfaktorer mens andre sekvenser ikke er det.
Slik som det ser ut foreløpig er alle sekvenser egentlig like sannsynlige.
Man kan derfor ikke bare lese seg gjennom en promotor-sekvens og si at
"dette området her ser ut som et fint motiv" (med mindre man da tilfeldigvis gjenkjenner
et motiv som man visste om fra før).
Som oftest benytter man seg av en mer indirekte metode for å oppdage motiver:
Man starter med en mengde promotor-sekvenser for ulike
gener hvor man på forhånd har prøvd å forsikre seg om at alle sekvensene inneholder de
samme bindingsmotivene (selv om disse ennå ikke er kjent). En måte å finne slike sekvens-sett på er å
identifisere co-regulerte gener ved hjelp av microarray-eksperimenter.
Dette er en teknikk som gjør det mulig å måle hvordan mange ulike gener uttrykkes under forskjellige eksperimentelle
forhold. Gener som alltid reguleres opp og ned etter samme mønster er sannsynligvis co-regulert, og
det er rimelig å anta at promotor-sekvensene til disse genene inneholder bindingsseter for de samme
transkripsjonsfaktorene. Når man så har funnet en slik gruppe med co-regulerte gener, kan man gjøre statistiske
analyser på promotor-regionene for å avdekke om noen mønstre forekommer oftere enn det man burde forvente
sammenlignet med om man hadde studert et helt tilfeldig utvalg med promotorer.
For eksempel, la oss si at man har funnet 10 co-regulerte gen og har hentet ut utdrag fra promotor-regionene
til disse genene fra en database (f.eks. sekvenser på 1000 nukleotider som ligger foran hvert gen).
Ved gjennomgang av datasettet finner man ut at "ordet" ACATTGC forekommer i alle disse promotor-sekvensene.
I et tilfeldig datasett av denne størrelsen (10000 bokstaver) vil man forvente at
dette ordet forekommer 0.61 ganger. Sannsynligheten for at ordet skal forekomme tilfeldig hele 10 ganger i et
slikt datasett er cirka 0.0000001%. Ut fra våre antagelser om at gen-settet er co-regulert og derfor burde inneholde
flere forekomster av et bestemt motiv, virker det ikke lengre som noen tilfeldighet at dette ordet opptrer mange ganger.
Snarere kan det være en god indikasjon på at ordet nettopp er et bindingsmotiv for en transkripsjonsfaktor som er med på
å regulere uttrykket til disse 10 genene.
Selv om det ovennevnte eksempelet er realistisk er likevel motivoppdaging sjelden like enkelt i praksis.
En rekke forhold er med på å forverre oppgaven betraktelig:
For det første: Å finne sett av sekvenser som inneholder de samme motivene er langt fra lett.
Selv om den vanligste fremgangsmåten er å benytte
co-regulerte gen-sett fra mikroarray eksperimenter, garanterer det ikke at sekvensene man velger ut inneholder de
sammen bindingsmotivene. Én ting er at det er svært vanskelig å tolke de dataene man får ut fra disse eksperimentene.
En annen ting er at gener som har lik oppførsel ikke nødvendigvis må være styrt av de samme faktorene. Det kan være mange
ulike systemer som er med på å regulere oppførselen slik at resultatet blir som det blir. Og selv om man tilfeldigvis skulle
være helt sikker på at en gruppe gener faktisk er styrt av de samme faktorene, er det ikke sikkert at de promotor-sekvensene man plukker
ut inneholder bindingssetene man leter etter (disse bindingssetene trenger nemlig ikke alltid å ligge i promotor-regionen
men kan ligge langt unna eller til og med ligge inni eller etter selve genet). I praksis er det derfor ofte slik at bare
en del av promotor-sekvensene man analyserer inneholder bindingssetene man ønsker seg.
Motiver kan ofte være veldig korte og promotor regioner er potensielt veldig lange.
Det er ikke engang sikkert at man vet hvor lange promotor-regionene egentlig er, siden det ikke akkurat er satt opp noen skilt i sekvensen som sier
når den slutter. For å være sikker på å få med de aktuelle bindingssetene bør man derfor velge en "så lang bit som mulig" når
man henter ut sekvenser fra databaser.
Men dette gjør igjen at ulike motiv-kandidater har større sjanse for å opptre rent tilfeldig i datasettet.
Biologiske sekvenser er stort sett ikke tilfeldige. Man kan derfor ikke sammenligne mot en helt tilfeldig
bakgrunn for å finne ut om et spesielt ord er overrepresentert i sekvensen. Der finnes mange forskjellige
sekvenser som opptrer oftere en forventet
i genomet uten at det nødvendigvis betyr at disse er bindingsseter for transkripsjonsfaktorer. Eksempelvis består
omtrent halvparten av det menneskelige genomet av ulike former for "repeterende elementer" som er kortere eller lengre
sekvensbiter som forekommer mange ganger (faktisk utgjøres hele 10% av
én eneste 300 nukleotid lang sekvens som kalles Alu og som forekommer i en million eksemplarer spredt rundt i genomet.)
Sist men ikke minst: Bindingsmotiver er nesten aldri konservert men kan variere fra et bindingssete til et annet.
For eksempel kan den samme transkripsjonsfaktoren binde til sekvensen ACATTGC på et sted og til AGATTAC på
et annet sted, slik at det faktiske bindingsmotivet heller bør sies å være "AxATTxC" hvor to av posisjonene kan variere. Ofte
kan alle posisjonene i et bindingsmotiv variere i større eller mindre grad!
Alle disse faktorene er med på å senke signal/støy-forholdet og gjør det vanskeligere å skille bindingsseter fra
bakgrunnen.
Det er ikke nødvendigvis slik at de
reelle bindingssekvensene opptrer så mye oftere enn det man burde forvente, slik at det bare er å sortere sekvenser etter hyppighet
og plukke fra toppen av bunken. Ofte forekommer sekvenser som ikke er motiver mye hyppigere enn motiver.
Bindingsmotiver skiller seg heller ikke ut fra resten av DNA-sekvensen på noen åpenlys måte, så det er tilnærmet umulig å se
forskjell på de delene av en sekvensen som faktisk er bindingsseter og de delene som ikke er det.
Å bedrive motivoppdaging i praksis kan derfor sammenlignes med å lete etter nåla i høystakken,
men i dette tilfellet er både høystakken og nåla laget av det samme materialet – DNA.
Selv om motivoppdaging er langt fra noen triviell oppgave har det likevel ikke manglet på forsøk opp igjennom årene.
Allerede er det blitt publisert over hundre ulike metoder.
I 2005 publiserte Tompa et al. en etterhvert meget berømt studie
i Nature Biotechnology
hvor de undersøkte og sammenlignet ytelsen til 13 av de mest brukte verktøyene for motivoppdaging på uavhengige datasett.
Resultatet var ganske nedslående. Ikke bare feilet programmene i å identifisere mange av de kjente bindingssetene i sekvensene,
men de predikerte også mange bindingsseter på steder hvor det antatt ikke skulle være noen. På reelle datasett
basert på det menneskelige genomet, presterte de fleste programmene ikke stort bedre enn det man kunne ha forventet om de
bare hadde gjettet helt tilfeldig.
Denne studien viste derfor at det fortsatt er et godt stykke igjen før vi kan si at vi har gode og robuste
verktøy for å finne bindingsseter for transkripsjonsfaktorer. En annen ting denne artikkelen
påpekte var at ulike programmer gjerne presterte forskjellig på ulike typer datasett. Mens for eksempel
et program gjorde det bra på noen bestemte datasett og dårlig på andre, kunne andre programmer igjen prestere
godt på de datasettene hvor det første programmet feilet. En måte å forbedre prediksjonsevnen på vil derfor
være å kombinere styrken fra flere ulike metoder.
Hvis du er interessert i å lese mer om motiver og motivoppdaging vil jeg foreslå
et par korte og gode introduksjonsartikler skrevet av Patrik D'haeseleer og publisert
i Nature Biotechnology:
What are sequence motifs? og
How does DNA sequence motif discovery work?.
<< Tilbake
|